
On this page
Key Takeaways
- IEC 81001-5-1 is the international standard for secure health software lifecycle activities, now cited by both FDA and EU regulators.
- The standard defines activities across the full lifecycle: development, verification, release, postmarket, and end-of-life.
- Security risk assessment under 81001-5-1 aligns to AAMI TIR57 and ISO 14971, the three form the anchor set for a Section 524B submission.
- Every activity produces objective evidence that lives inside the QMS under version control.
- The Feb 2026 FDA guidance explicitly names IEC 81001-5-1 as state of the art.
Learn how to implement IEC 81001-5-1 security risk assessments for FDA compliance. Expert guidance on medical device lifecycle security mapping.
This guide is written for medical device manufacturers navigating IEC 81001-5-1 security risk assessment. It is built from real submissions, FDA correspondence, and the standards reviewers actually cite. Use it as a working reference: read straight through, jump to the section that matches your current gap, or hand it to your engineering and regulatory leads as a checklist.
What is IEC 81001-5-1 and Why Does It Matter?
IEC 81001-5-1 is the international standard governing security activities across the full life cycle of health software and health IT systems, specifying what a manufacturer must do at each phase, from requirements and design through verification, release, and postmarket maintenance, to manage security risk. Unlike TIR57 or TIR97, which are AAMI technical information reports offering guidance, IEC 81001-5-1 is a normative international standard that can be cited for conformity assessment, and it is increasingly referenced by both the FDA and European notified bodies as objective evidence of a mature security development process. For manufacturers selling into both the U.S. and EU MDR markets, it functions as a common backbone that satisfies overlapping premarket expectations from both regulators.
The Shift from Safety Risk to Security Risk
Traditional medical device risk management, built around ISO 14971 and IEC 62304, was designed to manage risk from random failures and unintentional design defects, not intentional, adaptive threat actors. IEC 81001-5-1 formalizes the recognition that security threats behave differently: they evolve after release, they can be introduced by third-party software components you did not design, and they require ongoing monitoring rather than a one-time verification event. This shift means manufacturers need security-specific life cycle processes, secure coding standards, security testing, vulnerability monitoring, layered on top of, not replacing, their existing safety-focused quality processes.
Relationship with ISO 14971 and AAMI TIR57
IEC 81001-5-1 does not replace ISO 14971's safety risk management process, it operates alongside it, feeding security-relevant risk information into the same risk management file where a security exploit could produce patient harm. It shares significant conceptual overlap with AAMI TIR57, both describe a security risk management process modeled on ISO 14971's structure, but IEC 81001-5-1 is broader in scope, covering the full software life cycle process (aligned with IEC 62304) rather than only the risk assessment activity itself. Manufacturers already following TIR57 typically find that most of the underlying risk assessment work transfers directly, with additional life cycle process documentation needed to fully satisfy IEC 81001-5-1's structure.
Core Requirements of an IEC 81001-5-1 Security Risk Assessment
IEC 81001-5-1 requires manufacturers to define security requirements early in development, maintain a documented software life cycle that maps security activities to each phase (aligned with IEC 62304's life cycle model), and produce verifiable evidence that security risks were identified, evaluated, and controlled to an acceptable level before release. It also requires ongoing security update management and a defined process for handling vulnerabilities discovered post-release, extending the standard's reach into the postmarket phase rather than treating security as a premarket-only concern.
Security in the Product Lifecycle (SPDF)
The standard expects security activities to be embedded at every stage of the SPDF: security requirements elicitation during planning, threat modeling and architecture review during design, secure coding and static analysis during implementation, security-focused test cases during verification, and a defined process for security patch management and vulnerability monitoring after release. This lifecycle mapping is what distinguishes IEC 81001-5-1 from a standalone risk assessment exercise, it forces security to be a first-class part of every IEC 62304 life cycle phase rather than a parallel activity bolted on near the end.
Defining the System Boundary and Assets
Before assessing risk, IEC 81001-5-1 requires a clear definition of the system boundary, what is inside the manufacturer's control versus what belongs to the hospital network, cloud provider, or third-party component supplier, along with an inventory of the assets within that boundary: data, functions, credentials, and communication interfaces. Getting the boundary wrong is a common failure point, manufacturers who draw the boundary too narrowly (excluding, for example, a cloud analytics service they rely on) end up with a risk assessment that misses threats originating from components they assumed were "out of scope" but that still affect their device's security posture.
Step-by-Step Security Risk Management Process
Vulnerability Identification and Analysis
This step involves systematically identifying weaknesses in the device's design, implementation, and supporting infrastructure, using a combination of threat modeling, static and dynamic code analysis, SBOM-based known-vulnerability scanning, and manual security review of high-risk components such as authentication and update mechanisms. IEC 81001-5-1 expects this analysis to be repeated whenever the architecture or a key dependency changes, since a vulnerability introduced by a third-party library update is just as much in scope as one introduced by the manufacturer's own code.
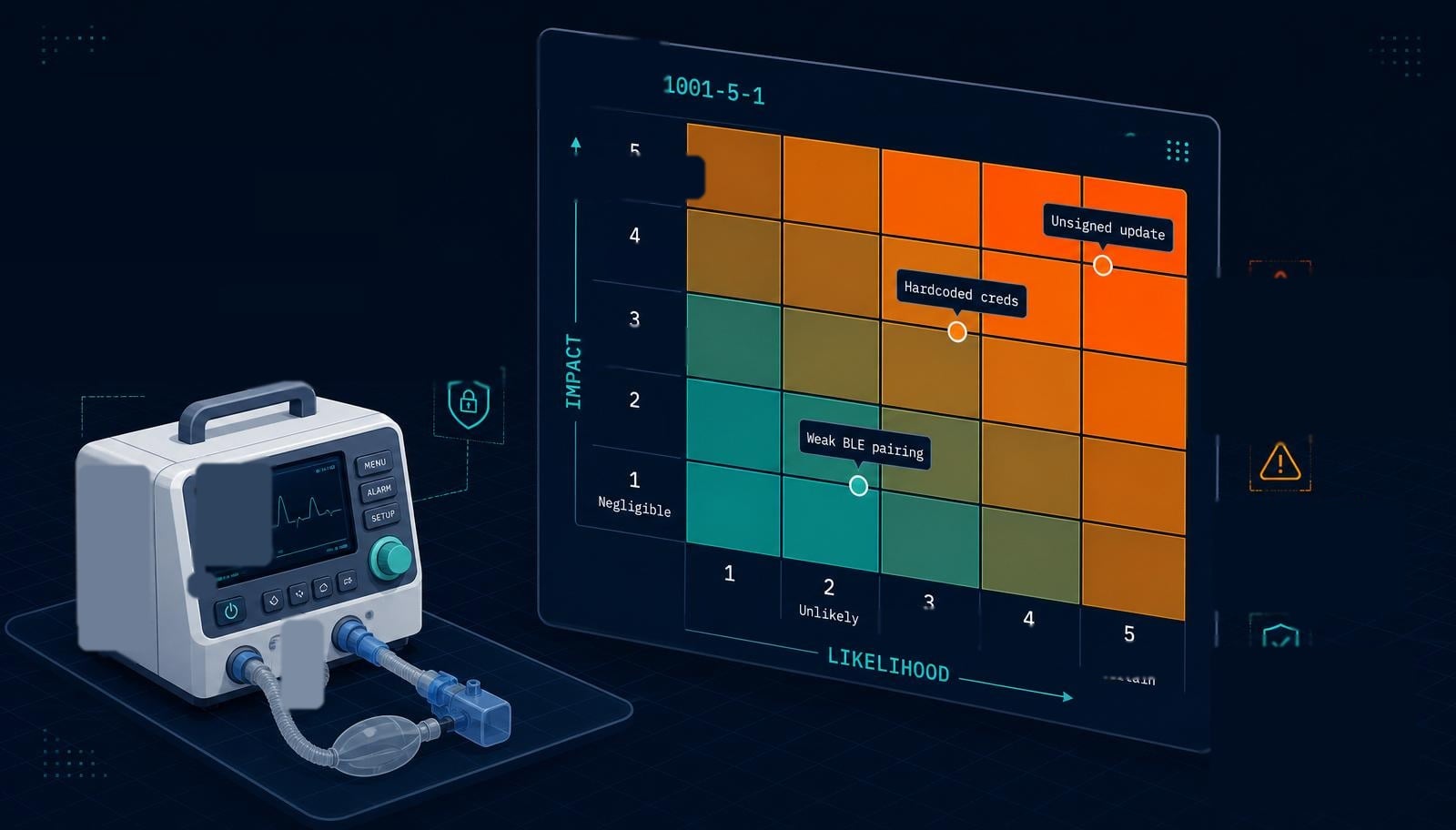
Risk Evaluation: Impact vs. Likelihood
Each identified vulnerability is evaluated against two dimensions: the potential impact if exploited (data breach, loss of device function, patient harm) and the likelihood of exploitation given the attacker skill, resources, and access required. IEC 81001-5-1 does not mandate a specific scoring methodology, but it does require the methodology to be documented, consistently applied, and defensible, many manufacturers adapt CVSS scoring or a custom matrix aligned with their ISO 14971 severity and probability scales so that security and safety risk can be compared on the same terms.
Control Implementation and Verification
Risks above the acceptability threshold require documented controls, encryption, authentication, input validation, network segmentation, or compensating procedural controls where a technical fix is not feasible, and each control needs verification evidence proving it actually reduces the risk as intended. This is where many assessments fall short in practice: a control listed in a spreadsheet without a corresponding test report demonstrating it works is not verification, it is an unverified claim, and reviewers increasingly ask for the underlying test evidence rather than accepting a narrative summary.
IEC 81001-5-1 vs. FDA Premarket Guidance
The FDA's premarket cybersecurity guidance does not require IEC 81001-5-1 conformance by name, but a manufacturer who follows the standard's life cycle process and produces its required documentation, security requirements, threat model, risk assessment, verification evidence, will satisfy the great majority of the FDA's substantive expectations for a cyber device submission. Because IEC 81001-5-1 is also recognized in the EU under MDR, building your process around it gives manufacturers targeting both markets a single security development process that supports two regulatory submissions instead of maintaining parallel, differently structured programs.
Mapping Standards to FDA Documentation Requirements
A practical mapping exercise ties each IEC 81001-5-1 life cycle activity to the specific FDA premarket submission artifact it supports: security requirements map to your cybersecurity risk assessment narrative, threat modeling output maps to your submitted threat model, and verification records map to your security testing summary. Building this mapping explicitly, ideally as a cross-reference table included in your submission, helps reviewers quickly confirm that your process addresses their expectations without having to reverse-engineer the connection themselves.
Addressing Security Risk in Section 524B Submissions
Section 524B requires manufacturers of cyber devices to have a plan for monitoring and addressing postmarket vulnerabilities, a process for providing reasonable assurance the device is cyber secure, and an SBOM, all of which align closely with what IEC 81001-5-1 already requires as part of its life cycle process. Manufacturers building their 524B compliance program directly on an IEC 81001-5-1-conformant process tend to have an easier time demonstrating each statutory element, since the standard already requires the underlying activities, monitoring, vulnerability management, documented risk control, that Section 524B is asking about.
Common Pitfalls in Security Risk Assessments
The most common failures we see are: drawing the system boundary too narrowly and missing third-party or cloud-hosted components that are still part of the attack surface, treating the risk assessment as a one-time document rather than a life cycle process that gets revisited at each design change, using an undocumented or inconsistent risk scoring methodology that reviewers cannot follow, and claiming controls are implemented without corresponding verification test evidence. Each of these produces an assessment that looks thorough on the surface but collapses under specific reviewer questions about a particular component or control.
Expert Support for IEC 81001-5-1 Compliance
Standing up a fully conformant IEC 81001-5-1 process from scratch, life cycle mapping, threat modeling, risk scoring methodology, verification test planning, is a significant undertaking for teams without dedicated security engineering resources, and getting the system boundary or risk methodology wrong early tends to compound into rework across every later phase. Manufacturers benefit most from bringing in security expertise during the requirements and architecture phases, when it is still cheap to change the design, rather than retrofitting the process just before a submission deadline.
How Blue Goat Cyber Approaches IEC 81001-5-1 security risk assessment
We treat IEC 81001-5-1 security risk assessment as a regulated engineering workstream, not a one-time document drop. Every engagement is led by senior medical-device security engineers who have shipped 250+ FDA cybersecurity submissions across 510(k), De Novo, PMA, and EU MDR pathways. Here is how we run it end to end:
- Scoping against your device profile. We baseline connectivity, interfaces, data flows, and intended use before we touch a template - because reviewer expectations for a Class II wearable are not the same as a networked hospital platform.
- Standards mapping in writing. Every deliverable is traced to the February 2026 FDA premarket cybersecurity guidance, AAMI SW96, AAMI TIR57 / TIR97, IEC 81001-5-1, and ISO 14971 - with the citation in the artifact itself so reviewers do not have to guess.
- Evidence generated inside your QMS. Threat models, SBOMs, security risk assessments, and test reports are versioned under design controls so the traceability from requirement → test → residual risk holds up under audit.
- Independent testing where it counts. Penetration testing and vulnerability analysis are executed by a testing team that does not also write the design - the separation FDA reviewers increasingly expect on cyber devices.
- Deficiency-ready posture. We anticipate the RTA, AI-letter, and Major deficiency patterns FDA has issued over the past 24 months and pre-empt them in the initial submission, cutting the odds of a second review cycle.
- Postmarket handoff, not abandonment. Every premarket package leaves you with a working postmarket monitoring plan, CVD process, and update cadence so the evidence you shipped stays defensible after clearance.
If you want that treatment applied to your IEC 81001-5-1 security risk assessment package, our FDA Premarket Cybersecurity Services and FDA Cybersecurity Deficiency Response engagements are the two most common entry points.
Frequently asked questions
How does IEC 81001-5-1 differ from ISO 14971?
ISO 14971 manages safety risk arising from any cause, random hardware failure, use error, or software defect, evaluated against harm to the patient, operator, or environment. IEC 81001-5-1 manages security risk specifically, arising from intentional threat actors exploiting vulnerabilities in health software and health IT systems, and it defines a full software life cycle process for security rather than just a risk evaluation methodology. The two standards are meant to work together: security risks identified under IEC 81001-5-1 that could produce patient harm should feed into the same ISO 14971 risk management file, so a reviewer sees one consolidated risk picture rather than two disconnected assessments.
Is IEC 81001-5-1 mandatory for FDA medical device submissions?
The FDA does not cite IEC 81001-5-1 as a mandatory standard by name in its premarket cybersecurity guidance, but the standard is increasingly recognized as objective evidence of a mature security development process, and its life cycle activities closely mirror what FDA reviewers expect to see documented regardless of which named standard you reference. For manufacturers also targeting the EU market under MDR, IEC 81001-5-1 carries more direct regulatory weight, and building a single process around it, rather than maintaining separate U.S. and EU-specific processes, is generally the more efficient path.
What are the specific security life cycle activities required by IEC 81001-5-1?
The standard requires security requirements elicitation during planning, threat modeling and architectural risk analysis during design, secure coding practices and static analysis during implementation, dedicated security test cases during verification, and a documented process for vulnerability monitoring, patch management, and disclosure during the maintenance and postmarket phase. Each activity needs to produce retained evidence, not just be performed informally, since the standard's conformity assessment relies on being able to demonstrate each life cycle phase was executed and documented as specified.
How do you integrate threat modeling into an IEC 81001-5-1 assessment?
Threat modeling, typically using STRIDE, sits inside the design phase of the IEC 81001-5-1 life cycle and produces the threat catalog that feeds directly into the standard's vulnerability identification and risk evaluation steps. Run the threat model against the system boundary and asset inventory you defined at the start of the assessment, score each identified threat using your documented risk methodology, and carry forward the resulting risk register into your control implementation and verification activities so there is a clear, traceable line from threat to mitigation to test evidence.
Does IEC 81001-5-1 apply to SaMD (Software as a Medical Device)?
Yes, IEC 81001-5-1 applies broadly to health software and health IT systems, which explicitly includes standalone Software as a Medical Device, not just software embedded in a physical device. SaMD manufacturers often find the standard particularly relevant because their system boundary frequently includes cloud infrastructure, mobile companion apps, and third-party APIs, exactly the kind of distributed architecture the standard's asset inventory and boundary definition steps are designed to force manufacturers to account for comprehensively.
Where this fits in the cluster
This page sits downstream of our pillar resources on IEC 81001-5-1 security risk assessment. If you arrived here from a different starting point, these are the most useful adjacent pages:
- FDA Premarket Cybersecurity Services
- The MedTech Cybersecurity Standards Decoder
- Secure MedTech Product Design Consulting
- The SPDF Playbook for FDA-Ready Medical Devices
Related from Blue Goat Cyber
- Medical Device Threat Modeling
- FDA-Compliant SBOM Services
- Medical Device Penetration Testing
- 12 Reasons the FDA Rejects Medical Device Cybersecurity Submissions
- The Postmarket Cybersecurity Readiness Plan
Sources & primary references
- Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions. FDA (U.S. Food and Drug Administration)
- IEC 81001-5-1:2021 - Health software and health IT systems safety, effectiveness and security. Part 5-1: Security. Activities in the product life cycle. ISO/IEC (International Organization for Standardization)
- AAMI TIR57: Principles for medical device security, Risk management. AAMI (Association for the Advancement of Medical Instrumentation)
- NIST SP 800-30 Rev. 1: Guide for Conducting Risk Assessments. NIST (National Institute of Standards and Technology)
Talk to a regulatory cybersecurity team
If you are working through IEC 81001-5-1 security risk assessment and want a second pair of eyes on your submission package, we ship cybersecurity deliverables for medical device manufacturers across 510(k), De Novo, PMA, and EU MDR pathways. Book a discovery session and we will walk your evidence with you.
Sources & references
Primary sources cited in this article. Links open in a new tab.
- Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions- U.S. FDA
- IEC 81001-5-1:2021 - Health software and health IT systems safety, effectiveness and security. Part 5-1: Security. Activities in the product life cycle- ISO
- AAMI TIR57: Principles for medical device security, Risk management- AAMI
- NIST SP 800-30 Rev. 1: Guide for Conducting Risk Assessments- NIST


