On this page
Key Takeaways
- Section 524B compliance requires three documented deliverables: postmarket vulnerability and patch plan, SPDF-aligned design process, and machine-readable SBOM.
- The FDA has RTA authority for missing artifacts, enforcement began October 1, 2023.
- Every artifact must cite the Feb 2026 final premarket guidance section it addresses.
- SBOMs must be CycloneDX 1.5+ or SPDX 2.3+ files, not PDFs.
- Postmarket plans must include CVD policy, monitoring cadence, and patch delivery, not just intent.
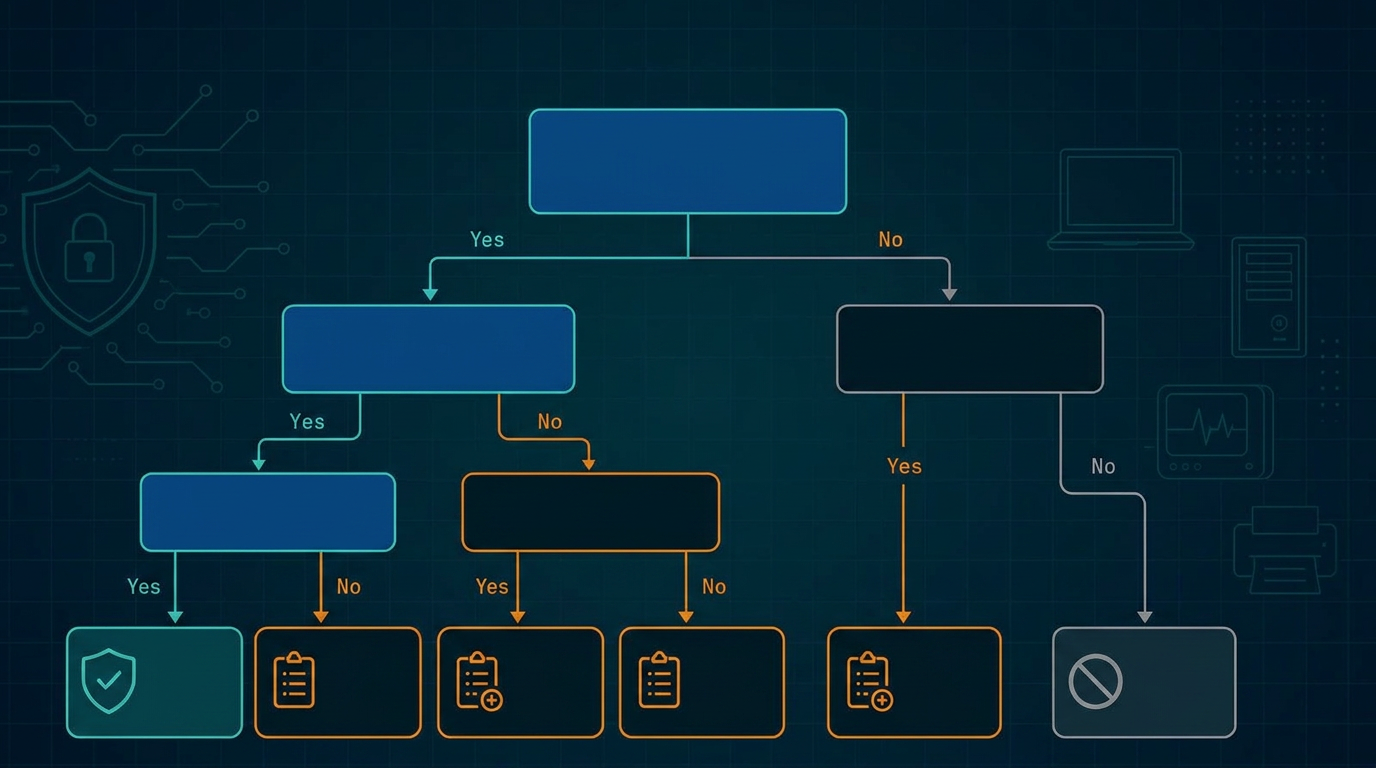
Use this checklist to confirm your premarket submission satisfies every statutory clause of Section 524B and the corresponding deliverable described in the FDA's February 3, 2026 final premarket cybersecurity guidance. Missing any item below is the most common reason cyber device submissions get a Refuse-to-Accept (RTA) hold or a Major deficiency letter.
Last reviewed: June 2026 against the FDA final guidance issued February 26, 2026.

Section 524B timeline
- December 29, 2022 - 524B became law, signed as part of the Consolidated Appropriations Act, 2023 (Pub. L. 117-328), the FDORA provisions, Section 3305.
- March 29, 2023 - The law took effect, 90 days later, per the effective-date clause in Section 3305(c) of Pub. L. 117-328. The FDA's refuse-to-accept authority attached on this date.
- October 1, 2023 - The FDA actually began issuing refuse-to-accept decisions based on 524B, per the FDA "Refuse to Accept Policy for Cyber Devices" guidance (March 30, 2023), which stated the agency would not issue RTAs based solely on 524B before October 1, 2023. This is the real start-of-rejections date.
Who This Checklist Is For
This checklist is for sponsors preparing a 510(k), De Novo, PMA, PDP, or HDE submission for a cyber device as defined in Section 524B(c): any device that (1) includes software validated, installed, or authorized by the sponsor, (2) has the ability to connect to the internet, and (3) contains technological characteristics that could be vulnerable to cybersecurity threats. (IDE submissions sit outside Section 524B(a) but are still subject to cybersecurity expectations under the FDA's February 3, 2026 final premarket guidance. BLA and IND submissions are outside 524B entirely, but the guidance still applies to the device constituent of a combination product - see combination products: when device cybersecurity content lands in a BLA or IND.)
If your device meets all three prongs, every item below applies. The FDA does not distinguish between Class II and Class III for Section 524B purposes, the statute applies uniformly.
Part 1, Section 524B(b)(1): Postmarket Vulnerability Plan
The statute requires sponsors to "submit to the Secretary a plan to monitor, identify, and address, as appropriate, in a reasonable time, postmarket cybersecurity vulnerabilities and exploits, including coordinated vulnerability disclosure and related procedures." (21 U.S.C. 360n-2(b)(1))
Checklist:
- Vulnerability monitoring procedure that names the data sources (NVD, CISA KEV, vendor advisories, your SBOM-driven CVE feed) and the cadence of review.
- Coordinated Vulnerability Disclosure (CVD) policy published at a stable URL on your corporate site, with a security contact, PGP key or secure form, and a stated safe-harbor.
- Triage SLA defining how quickly you assess, score (CVSS v3.1 or v4.0), and disposition each reported vulnerability.
- Remediation SLA distinguishing routine patches (regular cycle) from out-of-cycle critical patches.
- Customer communication plan describing how field notices, advisories, and CSAF/VEX documents are distributed.
- VEX issuance commitment tying each non-exploitable CVE in your SBOM to a documented VEX statement.
→ Related: Medical Device VDP & CVD Workflows, VEX Documents for Medical Devices.
Part 2, Section 524B(b)(2): Secure Product Development Framework
The statute requires sponsors to "design, develop, and maintain processes and procedures to provide a reasonable assurance that the device and related systems are cybersecure, and make available postmarket updates and patches to the device and related systems." (21 U.S.C. 360n-2(b)(2))
Checklist:
- SPDF policy document mapped to AAMI TIR57 / ANSI/AAMI SW96:2023, IEC 81001-5-1, or ISO/IEC 27034.
- Security risk management file under IEC 81001-5-1, distinct from your ISO 14971 safety risk file but cross-referenced to it.
- Threat model (STRIDE or equivalent) covering every trust boundary, with mitigations traced to design controls.
- Architecture views, global system view, multi-patient harm view, updateability view, as required by the February 2026 guidance.
- Penetration test report covering vulnerability chaining, boundary analysis, closed-box testing, and manual exploit verification.
- Cybersecurity testing summary documenting unit, integration, and system-level security verification.
- Updateability and patchability design evidence, signed updates, rollback protection, secure boot chain, and demonstrated update mechanism.
- Cybersecurity Bill of Materials labeling for end users (security-relevant features, recommended configurations, decommissioning instructions).
→ Related: Security Risk Assessment under IEC 81001-5-1, STRIDE Threat Modeling for Medical Devices, Penetration Testing for Medical Devices, 12 Critical Threat Modeling Gaps.
Part 3, Section 524B(b)(3): Software Bill of Materials
The statute requires sponsors to "provide to the Secretary a software bill of materials, including commercial, open-source, and off-the-shelf software components." (21 U.S.C. 360n-2(b)(3))
Checklist:
- Machine-readable SBOM in CycloneDX 1.5+ or SPDX 2.3+ format covering every layer of the runtime (firmware, OS, middleware, application, third-party libraries).
- Component identifiers include PURL (for OSS components) and CPE (for FDA postmarket CVE matching) wherever both exist.
- Cryptographic hashes for each component to support integrity verification.
- Supplier and license metadata for each component.
- SBOM generation procedure documenting whether SBOMs are produced from source, build, or binary analysis, and how often they are regenerated.
- Known-vulnerability summary at time of submission, with VEX statements explaining the status of each open CVE.
→ Related: SBOM for Medical Devices, CycloneDX vs SPDX, CPE vs PURL, SBOM Vulnerability Management.
Part 4, Section 524B(b)(4): "Other Requirements" the FDA Establishes
The statute gives the FDA authority to establish additional requirements by regulation (21 U.S.C. 360n-2(b)(4)). The February 2026 final guidance is not that (b)(4) rulemaking - the guidance is nonbinding and interprets the existing (b)(1)-(3) requirements rather than expanding them. Actual expansion of statutory obligations would take rulemaking. That said, the guidance is the operative interpretation reviewers use, so the premarket cybersecurity submission should also include:
- Cybersecurity Management Plan as a top-level submission document.
- Interoperability considerations documenting connected systems, protocols, and shared-responsibility boundaries.
- Total Product Life Cycle (TPLC) cybersecurity narrative spanning premarket through end-of-support.
- Labeling for cybersecurity including end-user security responsibilities and known residual risks.
- End-of-support plan with a defined sunset policy and customer transition path.
- Manufacturer Disclosure Statement for Medical Device Security (MDS2) if requested by your customers' health-delivery organizations.
→ Related: FDA Premarket Cybersecurity Submission Checklist, FDA PMA Cybersecurity Requirements, SaMD Cybersecurity Requirements.
Part 5, Common RTA and Deficiency Triggers
Even with all four parts covered, these recurring issues drive Major deficiency letters. Pre-flight them:
- SBOM is a PDF, not machine-readable.
- Threat model omits trust boundaries the architecture diagram shows.
- Penetration test report has no manual exploit verification, only an automated scanner output.
- CVD policy is referenced but not published at a stable, crawlable URL.
- Updateability claim is not backed by an actual demonstrated update mechanism in the verification record.
- Architecture views are missing the multi-patient harm view.
- No VEX statements accompany known CVEs in the SBOM.
→ See real examples: FDA Cybersecurity Deficiency Letter Examples.
How to Use This Checklist
- Walk the checklist in order, the statute imposes the order, and FDA reviewers read submissions in roughly this sequence.
- For each unchecked box, identify the owner (regulatory, engineering, security) and a target date before submission.
- Re-run the checklist 30 days before submission and again 7 days before, with each owner signing off.
If a single box is unchecked at submission, the submission is incomplete by definition, Section 524B is statutory, not advisory.
How Blue Goat Cyber Approaches Section 524B Compliance
Section 524B is the legal spine of every cyber-device submission - and the reference technical screeners return to when they issue deficiencies. We build every package against it explicitly. Here is the model.
- 524B(b) subsection mapping. Every deliverable is tied to the specific 524B(b) subsection it satisfies (SPDF plan, SBOM, postmarket monitoring, coordinated disclosure), with the citation in the artifact.
- eSTAR v7 alignment. Deliverables are named and formatted to fit the current subforms, not the 2023 layout.
- February 2026 guidance overlay. 524B tells you what is required; the 2026 guidance tells you what "adequate" looks like. We satisfy both in the same artifact set.
- RTA and technical-screening pre-check. Before submission we run the same checks reviewers run and close the last gaps.
- Postmarket commitments as first-class deliverables. Monitoring plan, CVD process, and update cadence shipped alongside the premarket package, not promised for later.
- Senior engineers on every engagement. The person building your 524B evidence set is the person who would defend it if a deficiency arrived.
Our FDA Premarket Cybersecurity Services engagement is the standard entry point.
FAQ
Does Section 524B apply to my Class II device?
Yes, if your device meets the three-prong definition in Section 524B(c), has software, connects to the internet, and has technological characteristics that could be vulnerable, Section 524B applies regardless of risk class. The FDA does not exempt Class II.
Is a PDF SBOM acceptable?
No. The February 2026 final guidance expects a machine-readable SBOM in CycloneDX or SPDX format. PDFs are commonly cited as a Major deficiency.
How is Section 524B different from the FDA's February 2026 cybersecurity guidance?
Section 524B is the underlying statute (signed December 2022, with FDA RTA authority effective March 29, 2023 and active enforcement beginning October 1, 2023). The FDA's premarket cybersecurity guidance is nonbinding but operative: the September 2023 final was the first operational interpretation, superseded by the June 2025 final, then superseded by the February 3, 2026 final, which is the document reviewers actually use today.
Do I need penetration testing for a 510(k)?
Yes, if your device is a cyber device under Section 524B(c). The pathway does not change the statutory obligation. See Penetration Testing for Medical Devices for what an FDA-acceptable test report contains.
Where do I publish a Coordinated Vulnerability Disclosure policy?
At a stable, crawlable URL on your corporate site (commonly /security or /.well-known/security.txt). Reviewers and security researchers both expect to find it without authentication.
Get Help Compiling Your Section 524B Evidence
Blue Goat Cyber compiles every artifact on this checklist for medical device sponsors, SBOM, threat model, SPDX/CycloneDX generation, penetration testing, VEX issuance, CVD policy, and the cybersecurity management plan that ties them together. We have submitted under Section 524B across 510(k), De Novo, and PMA pathways since the statute took effect.
→ See our FDA Premarket Cybersecurity Services.
Sources & references
Primary sources cited in this article. Links open in a new tab.